
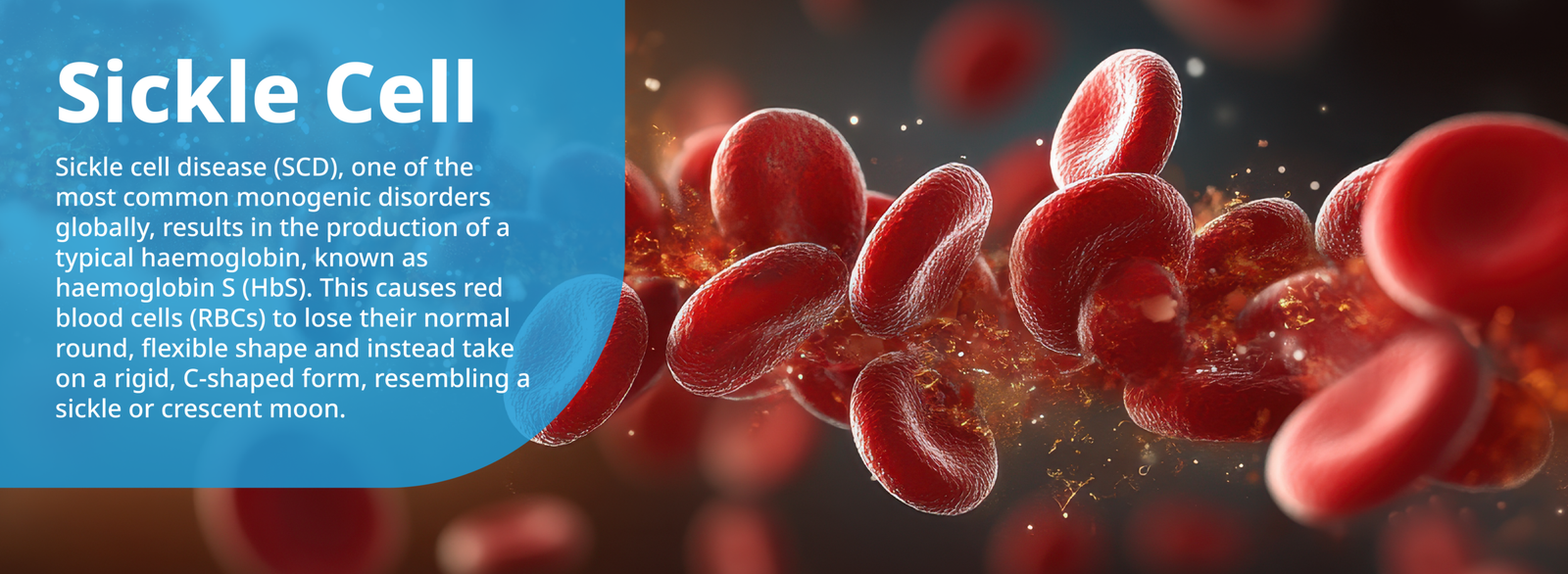
Sickle cell disease (SCD), one of the most common monogenic disorders globally, results in the production of a typical haemoglobin, known as haemoglobin S (HbS). This causes red blood cells (RBCs) to lose their normal round, flexible shape and instead take on a rigid, C-shaped form, resembling a sickle or crescent moon. These abnormal RBCs disrupt blood flow in small vessels and lead to blockages and inflammation, resulting in severe pain, organ damage and increased infection risk, thereby causing substantial morbidity and early mortality.1,2 Normal RBCs typically have a lifespan of ~120 days, but sickle cells only survive for 10 to 20 days, resulting in chronic anaemia due to the rapid destruction of these cells.2
A person can be a sickle cell carrier or have sickle cell disease if normal haemoglobin (HbA) is replaced by HbS. There are three common types of sickle cell diseases based on the haemoglobin genes inherited from the parents.2,3

SCD: Sickle Cell Disease, HbS: Haemoglobin S, HbA: Haemoglobin A, RBC: Red Blood Cells
SCD is highly prevalent in 17 states in India . It has a higher prevalence among the tribal population, with the Ministry of Health and Family Welfare recognizing this disease as one of the ten major health concerns disproportionately affecting these communities.2
A large-scale screening by the Ministry of Tribal Affairs, Indian Council of Medical Research, and Department of Biotechnology tested 113,83,664 individuals, identifying 996,368 (8.75%) as positive for SCD, with 949,057 carriers and 47,311 affected.2

SCD: Sickle Cell Disease, HbS: Haemoglobin S, RBC: Red Blood Cells

SCD is caused by a single-nucleotide polymorphism in the β-globin gene, substituting valine for glutamic acid at the sixth position. This mutation results in HbS, which polymerizes upon deoxygenation, causing RBCs to sickle. Sickling impairs blood flow (vaso-occlusion) and leads to ischemia-reperfusion injury. Additionally, HbS polymerization promotes haemolysis, releasing cell-free haemoglobin that depletes endothelial nitric oxide, thereby causing endothelial dysfunction. Reactive oxygen species and cell-free heme, a damage-associated molecular pattern, further promote sterile inflammation through inflammasome activation and IL-1β release. This exacerbates vaso-occlusion by increasing the adhesiveness of neutrophils, platelets, and endothelial cells, creating a cycle of vascular damage.4
IL-1β, Interleukin-1 beta

SCD is inherited in an autosomal recessive pattern. Individuals who inherit two copies of the mutated gene (one from each parent) develop the disease. Those who inherit one mutated gene and one normal gene have sickle cell trait and are generally asymptomatic but can pass the gene to their offspring.5

· Populations of Middle Eastern, Asian, Indian and Mediterranean descent
· Individuals of African ancestry, including African Americans
· Hispanic populations originating from Central and South America

A family history of SCD or sickle cell trait is a significant risk factor for developing the condition.7

· Dehydration, hypoxia, and extreme temperatures can trigger sickling episodes.9
· High altitude can exacerbate hypoxia-induced sickling.9,10
· Bacterial infections are a major cause of morbidity and mortality in SCD.11
SCD: Sickle Cell Disease
A Punnett square depiction of different combinations of parents’ genetic status and the probability of the children getting affected with SCD is as below2:

SCD: Sickle Cell Disease
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323. doi:10.1016/S0140-6736(17)30193-9
- National Sickle Cell Anaemia Elimination Mission 2023. Guidelines for national programme for prevention & management of sickle cell disease. Ministry Of Health & Family Welfare - Government of India; 2023. Accessed March 19, 2025. https://sickle.nhm.gov.in/uploads/english/OperationalGuidelines.pdf
- National Sickle Cell Anaemia Elimination Mission 2023. Training Module for Community Health Officers. Ministry Of Health & Family Welfare - Government of India; 2023. Accessed March 19, 2025. https://nhsrcindia.org/sites/default/files/2023-07/Training%20Manual%20on%20SCD%20for%20Community%20Health%20Officer%20%282%29.pdf
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14:263-292.
- Elendu C, Amaechi DC, Alakwe-Ojimba CE, et al. Understanding sickle cell disease: causes, symptoms, and treatment options. Medicine (Baltimore). 2023;102(38):e35237.
- Sickle cell disease. American Society of Hematology. Accessed March 19, 2025. https://www.hematology.org/education/patients/anemia/sickle-cell-disease
- Sickle cell disease - causes and risk factors. National Heart, Lung, and Blood Institute. 2024. Accessed March 19, 2025. https://www.nhlbi.nih.gov/health/sickle-cell-disease/causes
- Living with sickle cell disease. National Health Service (NHS). 2017. Reviewed November 30, 2022. Accessed March 19, 2025. https://www.nhs.uk/conditions/sickle-cell-disease/living-with/
- Tewari S, Brousse V, Piel FB, Menzel S, Rees DC. Environmental determinants of severity in sickle cell disease. Haematologica. 2015;100(9):1108-1116.
- Obadina M, Morris S, Alin T, LeVarge B, Little JA. High-altitude hypoxia is common in adults with sickle cell disease. Blood. 2023;142:3894.
- Sahu T, Pande B, Verma HK, et al. Infection and potential challenge of childhood mortality in sickle cell disease: a comprehensive review of the literature from a global perspective. Thalass Rep. 2023;13(3):206-229.